システム解析学分野
概要
多種多様な大規模生体データに対するデータ解析手法を開発し、がんの複雑なシステムの理解を進め、得られた知見を医療へ還元することを目指しています。
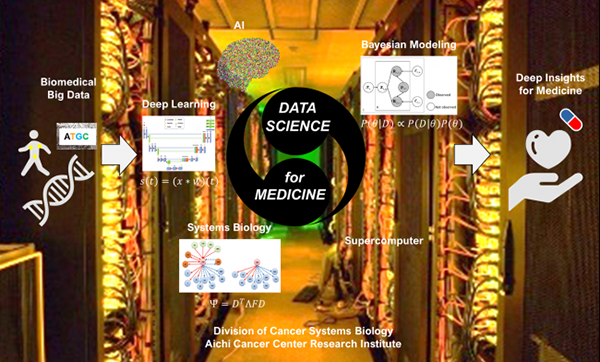
システム解析学分野では、ゲノム情報等の巨大パーソナルオミクスプロファイルデータの解析を通じて、がんの複雑なシステムを理解し、得られた知見を、個人に最適な予防や治療等に役立てることを目指して研究を行っています。そのために、深層学習モデリングやベイズ統計モデリング等の、先進的なデータ科学技術に基づくデータ解析手法を開発し、スーパーコンピュータを用いた解析にフル活用しています。更に、がんゲノム医療の現場で喫緊の課題となっている、エキスパートパネルにおける医療者の判断の精緻化・省力化に資する人工知能を活用した情報解析基盤技術の開発の研究も進めています。
目指すもの
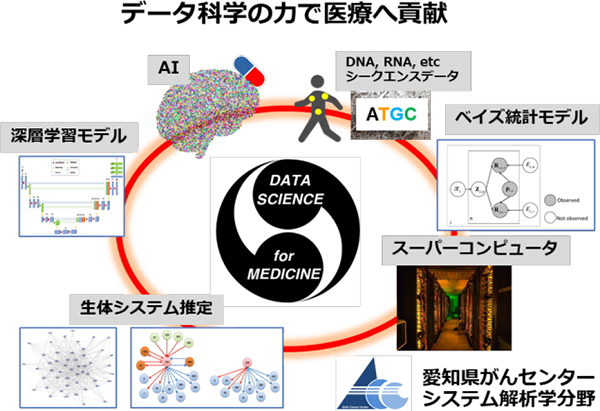
当分野のモットーは、“Data Science for Medicine”です。データ科学の力で医療へ貢献し、一人でも多くの患者さんの役に立てることを目指しています。 また医学、生命科学の分野は、今後ますますデータ科学、情報科学の力を身に着けた人材が必要とされ、活躍できる領域です。当分野が、そのような人材のインキュベーターとなることも目指しています。
連絡先
愛知県がんセンター研究所
システム解析学分野 分野長
山口 類(やまぐち るい)
〒464-8681 名古屋市千種区鹿子殿1-1
Tel:052-762-6111(内線)7310
E-mail:r.yamaguchi@aichi-cc.jp
研究内容 一般の方へ
はじめに
がん細胞は、正常な細胞中のDNA(ゲノム)配列に変異が起きることにより、細胞が異常に増殖する能力を獲得したものです。体の同じ臓器にできるがんであっても、ゲノム上で変異の起きる場所や変異の頻度は様々で、個人によっても違いがあります。私たちシステム解析学分野では、がん細胞のゲノム上の変異を、スーパーコンピュータを用いてできるだけ正確に見つけ出す先進的なデータ解析手法の研究を行っています。そして実際に得られた患者さんのデータからがんに関係する情報を抽出し、人工知能(AI)を活用して、短時間で正確に個人に最適な予防法や治療法を示すことができる情報解析技術の研究開発も行っています。
ベイズ統計モデリングに基づく高精度の変異検出技術の開発
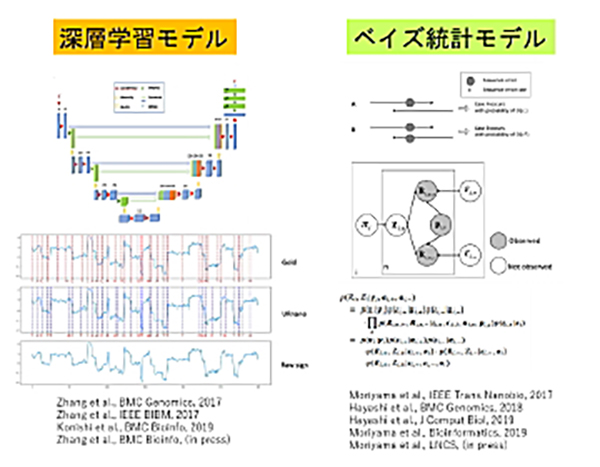
ヒト一人分のゲノムは4種類の塩基(A,T,G,C)からなる塩基の対30億個分からなります。ゲノム配列の解読は、シークエンサーと呼ばれる測定装置で行われ、例えるならばA、T、G、Cの4文字からなる30億文字分の文章が書かれた書類の束をシュレッダーにかけ、100文字ずつの断片として読み取るようなものです。シークエンサーでの読み取りでは一定の割合でエラーが生じるため同じものを30~40回読み込み、情報量はさらに多くなります。その膨大な文字片を今度はスーパーコンピュータでヒトのゲノムの標準的な配列と照らし合わせて並べ替え、どこにがん細胞特有の変異があるのかを探し出します。調べる組織の中にがん細胞が少ない場合などには、実際のがんの変異か、データの読み取りエラーかを見分けることが一層難しくなります。私たちシステム解析学分野ではベイズ統計モデルに基づいて、データの中に含まれる様々な補助情報を活用し、変異とエラーを高精度に見分ける情報解析技術の開発を行っています。
深層学習(ディープラーニング)技術に基づくデータ解析技術の開発
シークエンサーの飛躍的な性能の向上により、ゲノムの解読に要する費用と時間は劇的に圧縮され、治療の現場でも使うことができるようになってきました。最近はDNAを100文字の断片よりももっと長く読み取る「ロングリード」の技術が開発され、活用され始めました。DNAを長く一度に読み取ることができることで、ゲノム上の大規模な構造変異の検出に威力を発揮したり、同じパターンの配列が繰り返し現れる領域で正確に変異を検出したりする利点があります。一方で全般的には既存のシークエンサーによる読み取りに比べ正確性が低いことが問題となっています。当研究室では神経回路網を模倣して数式化したニューラルネットワークモデルを用いて、データに含まれる特徴を段階的に深く学習していく深層学習(ディープラーニング)の手法を用いて、読み取ったデータをA、T、G、Cの塩基の配列に正しく変換していく新たな解析手法を開発し、これまでより高い精度で塩基へ変換することを可能にしています。
人工知能(AI)によるがんゲノム情報の解析技術の開発
2019年、がんに関連する遺伝子の変異の調べる「がんゲノムパネル検査」が保険適用となりました。患者さん個人の遺伝子変異をもとに個別の医療をおこなう「がんゲノム医療」が本格化しつつあります。がんゲノム医療では、医師や看護師、薬剤師などが参加して「エキスパートパネル」という会議を開き、パネル検査の結果をもとに、患者さんの治療歴や健康状態など様々な状況を考慮して、どの治療法が適切かを検討します。このゲノム医療における課題の一つは、ゲノムの解析結果から得られる患者さんの大量の遺伝子情報の中から、治療のターゲットとなりうる変異の情報や、薬の効果を左右する要因となる情報などを、迅速かつ正確に読み解き治療につなげることです。私たちは人工知能を活用してパネル検査の結果を読み解くシステムの研究を進め、エキスパートパネルでの治療方針の決定の精緻化に役立つ情報解析技術の開発を行っています。
目指すもの
私たちの分野のモットーは“Data Science for Medicine”です。データ科学の力で医療に貢献し、一人でも多くの患者さんの治療に役立つことを目指しています。私たちは、愛知県がんセンター病院のエキスパートパネルへも参加し、治療現場からのフィードバックを活用して、DNAの情報だけでなく、将来はRNAやタンパク質、免疫情報などさらに膨大なデータを読み取り、素早く解析し、治療や診断に有用な情報に解釈して、患者さん一人ひとりにより適した治療の選択につなげられるシステムの開発を進めていく予定です。
また医学・生命科学の分野は今後ますますデータ科学、情報科学の力を身に着けた人材が必要とされ、活躍すべき領域です。当分野では、そのような人材の育成に積極的に寄与していくことも目指しています。
研究内容 専門家の方へ
概要
システム解析学分野は、がんの複雑なシステムをゲノム情報等の巨大パーソナルオミクスプロファイルデータの解析を通じて理解し、得られた知見を、個人に最適な予防や治療等に役立てることを目指して研究を行っています。そのために、深層学習モデリングやベイズ統計モデリング等の、先進的なデータ科学技術に基づくデータ解析手法を開発し、スーパーコンピュータを用いた解析にフル活用しています。また基礎的なデータ解析手法の開発に加え、がんゲノム医療の現場で喫緊の課題となっている、エキスパートパネルにおける医療者の判断の精緻化・省力化に資する人工知能を活用した情報解析基盤技術の開発を進めています。
1)ベイズ統計モデリングに基づく高精度体細胞変異検出技術の開発
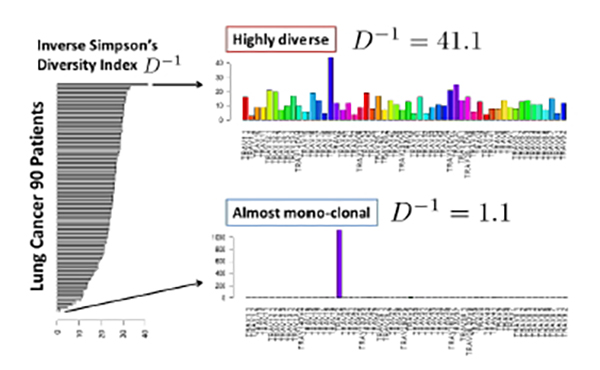
DNAシークエンスリードデータからの、体細胞変異検出(変異コール)において、リード中に含まれる読み取りエラーと真の変異情報を分離し、高精度に変異を検出するための、ベイズ統計モデリング技術に基づく手法を開発しています。このような高精度変異検出技術は、全ゲノムシークエンスデータのように、シークエンス読み取り深度の浅いデータからの変異検出や、低頻度変異アレル情報により規定される、がん細胞クローン集団構造の決定ために重要です。
上記の目的のために、従来の変異検出プロセスにおいて見逃されてきた、リード中に含まれる補助的な情報を活用する手法を研究してきました。例えば、あるペアードシークエンスリードの組が、同じDNAフラグメントを重複して計測した領域における変異候補パターンの情報を用いて、エラーと変異を高精度に分離する階層型ベイズモデル(OVarCall; Moriyama et al., 2017)を開発しました。また、他の種類の補助的情報を活用した変異検出モデルが複数存在する場合に、それらのモデル群からの情報を合理的に統合するベイズ統計的枠組み(OHVarfinDer; Moriyama et al., 2019a)を開発し、更なる変異検出精度の向上を示しました。また同一個人の複数領域シークエンスデータ中の変異候補情報を統合する、ベイズ型変異検出モデル(MultiMuc; Moriyama et al., 2019b)を開発し、各検体における変異検出の精度向上に成功しています。
2)免疫細胞シークエンスデータ解析技術の開発
がんと免疫の関係を解き明かし、治療に役だてることは近年のがん医療における大きな課題です。そのために、がんと免疫に関わる様々なモダリティを持つデータから有用情報を抽出するための手法群を開発しています。
一例として、T細胞受容体(TCR)やB細胞受容体(BCR)配列で特徴づけられる、免疫細胞クローンレパトア解析のためのシークエンスデータ解析パイプライン(TCRip/BCRip; Fang, Yamaguchi, et al., 2014他)を開発しています。ここではTCRまたはBCRのRNAシークエンスデータから、リードの部分配列アライメントを元に、高精度に各クローンを特徴づける配列を決定し、レパトア構造を推定するアルゴリズムを考案しています。同パイプラインは、がんワクチンや免疫チェックポイント阻害剤治療前後のレパトア構造の動的変化の解析等に活用されています。
また他に、DNAシークエンスデータからのHLA遺伝子型を高精度に決定する、階層型ベイズモデル(ALPHLARD; Hayashi et al., 2018)の開発を行っています。本手法は、読み取り深度が浅い全ゲノムシークエンスデータにおいても、既存の手法を大幅に上回る精度で、HLAの型決定を行うことが出来ます。また他の多くの手法において、型決定を行うことができないHLA クラス ll 伝子の型決定も行うことができます。同手法を用いて、決定されたHLA型情報は、ICGC Pacncancer Analysis of Whole Genomes(PCAWG)Project内の共通リソースとして活用されています。また、同手法を改良し、がん細胞、正常細胞のペアのシークエンスデータの情報を同時に考慮するモデル(ALPHLARD-NT; Hayashi et al., 2019)を開発しています、その結果、更なるHLA遺伝子型決定の精度の向上を得るとともに、HLA遺伝子中の体細胞変異の高精度検出にも成功しています。
3)深層学習技術に基づくシークエンスデータ解析技術の開発
人間の先験的知識によるモデル化や特徴抽出が困難な問題において、深層学習モデルは有効です。我々はシークエンスデータからの情報抽出において、深層ニューラルネットワークモデルを活用した手法を開発研究してきています。例えば、RNA-seqデータに含まれる様々なバイアスの補正にリカレントニューラルネットワークを用いた手法を考案しています(Zhang et al., 2017a)。コピー数変異などの大きな構造変異を高速かつ高精度に決定するためのモデルも研究しています(Zhang et al., 2017b)。
また、ナノポアシークエンサーからのロングリードが活用され始めていますが、既存のショートリードシークエンサーからのデータに比べて正確性が低いことが問題となっています。これは、ナノポアをDNAストランドが通過するときに検出される電流の値を、正確にDNAの塩基配列に変換できないからです。我々は、この問題に対して、新たな深層ニューラルネットワークモデル(URnano; Zhang et al., (in press))を提案し、既存の手法によりも高い精度で塩基への変換を果たしています。
4)がんゲノム臨床シークエンスのための情報解析基盤技術の開発
がんゲノムパネル検査が2019年より保険適用となり、がんゲノム医療が本格化しようとしています。我々は、近未来の、全ゲノムシークエンスおよび複数オミックスデータの統合解析に基づく、がん臨床シークエンスの実現に向けた情報解析基盤技術の開発を進めています。
臨床シークエンスにおいて喫緊の課題は、高速のデータ解析に加え、ゲノム解析の結果得られた変異情報を臨床上有用な情報へ、迅速かつ正確に解釈・翻訳し、エキスパートパネルにおける意思決定の精緻化につなげることです。そのために人工知能を活用した解釈システムの研究を進めています。当分野は、愛知県がんセンター病院のエキスパートパネルへも参画しており。今後、現場からのフィードバックを活用して、より実践的かつ未来を見据えたシステムの開発を進めていく予定です。
スタッフ紹介
計測技術の急速な発展とともに、これまでに手にすることのできなかった多種多様な生体データが産生されてきています。これらの情報の海の中から、これまで知り得なかった有用な情報を抽出し、現在および近未来の医療へ繋げることが喫緊の課題です。そのためには、データ科学の力が不可欠です。共にこの課題に取り組む、研究員、リサーチレジデント、大学院生を募集しています。
業績
リサーチレジデント募集
データ科学の力でがんのシステムの理解を進め、医療・生命科学へ貢献することに興味のある方、研究員及びリサーチレジデントの方を募集しております。また名古屋大学大学院医学研究科の連携大学院生として研究することも可能です。
